HP-model
En HP-model er den simpleste model, der kan beskrive proteinfoldning. Et protein er en fleksibel polymer bestående af flere aminosyrerester, der interagerer med hinanden og med omgivelserne for derved at give proteinet en bestemt udformning kaldet en foldning. Der findes 20 forskellige slags aminosyrer, men i HP-modellen forsimples disse til blot to typer aminosyrer - hydrofobe (H) og polære (P). Systemet simplificeres yderligere ved, at proteinet kun kan eksistere på et diskret gitter - et gitterprotein. I en vandig opløsning vil den mest fordelagtige proteinfoldning da være den, hvor færrest mulige H-aminosyrer er i kontakt med solventet og P-aminosyrerne.[1]
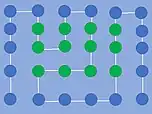
En termodynamisk stabil konformation i HP-modellen, hvor de grønne aminosyrer er hydrofobe. Der er 14 nærmeste-nabo-interaktioner imellem H- og P-aminosyrerne.
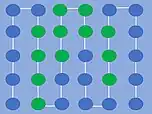
En termodynamisk ustabil konformation. Der er i alt 19 H-P-interaktioner plus 3 interaktioner i kanterne, hvor H-aminosyrerne er i kontakt med det polære solvent.
HP-modellen betragter forskellige mulige konformationer for at se, om de er stabile.[1]
Kildehenvisninger
- Phillips, Rob; Kondev, Jane; Theriot, Julie; Garcia, Hernan G. (2003). "8.4 - Proteins as Random Walks". Physical Biology of the Cell (engelsk) (2. udgave). Garland Science. s. 344-351. ISBN 978-0-8153-4450-6.
- Dill K.A. (1985). "Theory for the folding and stability of globular proteins". Biochemistry. 24 (6): 1501-9. doi:10.1021/bi00327a032. PMID 3986190.
| | Spire Denne artikel om fysik er en spire som bør udbygges. Du er velkommen til at hjælpe Wikipedia ved at udvide den. |
This article is issued from Wikipedia. The text is licensed under Creative Commons - Attribution - Sharealike. Additional terms may apply for the media files.